On December 3, 2024 we received word that the very first person received a new drug, called ALN-HTT02, as part of a Phase 1 trial aiming to treat Huntington’s disease (HD). Going boldly as the first human ever to receive this drug, they’re charting a course that we all hope will one day lead to a disease-modifying drug for HD. Let’s dig into the details of this new trial!
Who is behind this new trial?
In early November, Alnylam Pharmaceuticals announced that they were launching a clinical trial to test their huntingtin-lowering drug. A spelling error in the gene huntingtin, called HTT for short, is what causes HD. Those who inherit this spelling mistake in HTT will go on to develop changes with mood, movement, and memory as HD progresses. Lowering levels of the HTT molecule with the spelling mistake is one strategy being tested in the clinic to potentially treat HD.
Alnylam is a relative newcomer to the scene of HD therapeutics, but they’re not new to drug discovery. They’ve been around for 22 years and received commercial approval for their first drug 6 years ago. They also have experience with other brain disorders; they’re currently advancing towards a Phase 2 trial for a drug that they hope will treat Alzheimer’s disease.
The company is named for the bright center star in the constellation Orion’s belt, “Alnilam”. As that star has been used for thousands of years by navigators, it symbolizes Alnylam’s passion for discovery. For some, this star represents a bridge between earthly and celestial realms. Hopefully all these positive cosmic analogies are good luck and Alnylam’s drugs can bridge us to an HD-free future!
Molecular Mars Rovers
The drug that Alnylam developed in collaboration with Regeneron Pharmaceuticals that is being tested in this trial is called ALN-HTT02. It works through a mechanism called “RNA interference”, also known as RNAi. RNAi is unique because it takes advantage of molecular machinery that already exists inside cells for processing – like little Mars Rovers processing samples from the red planet. RNAi is Alnyam’s specialty. They developed the world’s first RNAi-based medicine! (A drug used to treat a nerve disease.)
ALN-HTT02 itself is a piece of synthetic genetic material that contains part of the code for HTT. Once it’s introduced to a cell, the cell’s own molecular machines are used to process the synthetic genetic material. This creates a fragment of the synthetic genetic material that binds to the HTT message to lower it.
ALN-HTT02 targets the first bit of the HTT gene called “exon 1”. This is the part of the genetic message that contains the spelling error that causes HD. Researchers think that exon 1 could be the key to driving toxicity that builds up over time, damaging brain cells. Hopefully by specifically targeting this area, that damage will be lessened, or stopped altogether.
Same twinkle, different star
Other ongoing clinical trials are taking a similar approach – targeting HTT to lower levels. While there are similarities to these other trials, there are also some subtle differences.
Like most other trials, ALN-HTT02 is what we call “total HTT” lowering. That means it targets both copies of the HTT gene, the one from mom and the other from dad. This means that both regular HTT and the HTT with the spelling mistake are lowered. Other companies, like Wave Life Sciences and Vico Therapeutics, are running trials that specifically or preferentially target only the copy of HTT with the disease-causing spelling mistake.
There are currently quite a few HTT lowering drugs being tested in clinical trials, but the way the drugs actually lower HTT levels differs. Companies like Wave, Vico, and Roche are using something called an antisense oligonucleotide, or an ASO. This is a short piece of genetic material that binds the HTT message molecule to lower levels. So it doesn’t use the cell’s molecular machinery to produce the message-binding fragment the way that RNAi does.
Companies like PTC Therapeutics and Skyhawk Therapeutics are using something called splice modulators that target HTT to lower levels. These are small molecules taken as a pill that target the way the HTT message molecule is processed, causing it to be sent to the cell’s trash can, like the waste containment units on the International Space Station. (Clearly there are only so many relevant space analogies…)
Similar to Alnylam, uniQure is using an RNAi-based approach. The difference between uniQure and Alynlaym though is that uniQure’s drug AMT-130 is carried in a harmless virus and delivered via brain surgery. Conversely, Alynalym’s ALN-HTT02 isn’t encapsulated in a virus and is delivered via spinal injection.
Overall though, all of the drugs being tested by these companies have the same goal – lower the HTT message to hopefully reduce the toxic effects of the protein with the goal of slowing or stopping HD.
Details about the trial
ALN-HTT02 is being tested in Phase 1 trial. As with all Phase 1 trials, the primary goal of this study will be to determine if it is safe. Phase 1 trials are the first time that drugs developed in the lab are ever given to humans! Knowing that they’re safe and well tolerated by people is the first step in advancing them in the clinic.
They’ll also look at how well the drug targets HTT and how levels change in the CSF, the fluid that bathes the brain. They’ll use clinical tests to measure symptoms, but would need a larger trial with different measures to understand if ALN-HTT02 works to change clinical features of HD.
Up to 54 people with Stage 2 or early Stage 3 HD between the ages of 25 and 70 are being recruited for this trial. Recruitment age is notable here, as most trials exclude those over the age of 65.
Currently, recruitment is only open at two sites in the UK, but the study is also being initiated in Canada and recruitment in additional countries is expected to follow.
Participants will be given a single dose of ALN-HTT02 via spinal injection during the trial. A portion of the participants will be given a placebo, an injection that contains no active drug. After 12 months, those who received the placebo will be given the option to receive an injection that contains ALN-HTT02.
Guided by the stars
As our first trial participant steps boldly into a future unknown, guided by a company that is inspired by the stars, we hope this charts a new path for HD therapeutics. Phase 1 trial participants are incredibly brave, like astronauts walking courageously into the unknown.
We are profoundly grateful to these brave volunteers who help answer questions about new drugs, taking the first steps across the celestial bridge to a future without HD.
Here, we cover a report on Advanced Care Planning (ACP) prepared by clinical researchers at the University College London, reminding us there are things that can be done right now to improve the lives of people living with HD. Until we have drugs that slow the progression of HD, peace of mind can be some of the best medicine.
Facing the challenges of HD doesn’t have to be done alone
Anyone dealing with Huntington disease (HD) knows that it comes with many physical and mental challenges, and these can affect everyone differently. For some, symptoms may eventually call for life changes such as moving to a long-term care facility.
As the ability to make decisions can be lost in the later stages of HD, it can help to express care preferences early on to ensure that one’s wishes are respected. Because each person’s journey with HD is unique, individualized and tailored care plans are crucial.
The best way to navigate these challenges is by openly discussing preferences with loved ones and healthcare providers. To support this, a team of clinicians have published a guide to help individuals and families plan ahead, empowering people with HD to retain a sense of autonomy and extend their independence, even after they can no longer make decisions independently.
What is Advanced Care Planning?
ACP is a process that helps individuals reflect on, understand, and communicate their concerns, preferences, and wishes for future medical care. While thinking about future health decisions can feel overwhelming, many people find value in discussing these matters, once they are ready.
Although doctors might have concerns that these conversations may cause undue stress, studies have shown that many individuals appreciate the chance to talk about their future care, as long as it is done in a personalized and context-specific way.
ACP can empower people with HD and extend their autonomy by ensuring that their choices are respected even when they may no longer be able to communicate them. It also reassures healthcare providers that the care they deliver aligns with a person’s values and wishes. For caregivers, knowing a person’s preferences can reduce feelings of helplessness, as they can act in ways that truly honor the individual. Documenting these preferences can even support legal aspects of care, such as appointing a power of attorney.
A roadmap for Advanced Care Planning
Previous studies have found that ACP is often underutilized in clinical practice, despite its benefits. Doctors have to approach ACP with sensitivity, recognizing that not everyone may be ready or willing to discuss future care decisions, while balancing this sensitivity with the benefits gained from setting things up in advance.
Addressing these challenges, a team of clinical researchers at the Huntington’s Disease Centre at University College London have developed a system to offer ACP to all patients who express interest. Their approach ensures that each person with HD has the opportunity to initiate ACP discussions whenever they feel ready.
People don’t need to make every decision at once when beginning ACP. Initial conversations can focus on exploring goals and values, providing a foundation for more specific plans later on. ACP is an ongoing, flexible process that accommodates changing views–these discussions are documented and reviewed regularly to reflect any updates in a person’s preferences.
Putting it on the record
In the case of the Huntington’s Disease Centre at University College London, ACP can eventually lead to some key documents. An Advance Statement captures personal wishes and preferences for end-of-life care or when decision-making capacity is lost. While not legally binding, it guides future care decisions and can include things like religious beliefs, preferred care locations, opinions on certain treatments, and funeral preferences.
Another option is an Advance Decision to Refuse Treatment (ADRT), a legally binding document where an individual can specify refusal of certain treatments, like artificial ventilation or resuscitation, if they lose capacity.
A Lasting Power of Attorney (LPA) allows a trusted person to make decisions on the individual’s behalf if they become unable to do so themselves. Given that each country has different regulations, it is important to discuss options with a healthcare team. This report offers a framework for healthcare teams to support ACP, helping to personalize and honor people’s wishes across different countries and settings.
What can be done now?
A Chinese proverb goes, “The best time to plant a tree was 20 years ago. The second-best time is now.” Similarly, the report highlights that while many people delay starting ACP and can still benefit from it, starting earlier is also an option.
HD is a long journey, allowing people ample time to approach ACP thoughtfully, ensuring that they’re ready to engage in these discussions. Recognizing that decisions don’t have to be made all at once and progressing at a comfortable pace can offer peace of mind.
In the spirit of gratitude, this Giving Tuesday, the HDBuzz editorial team would like to reflect on the unique and transformative role the Huntington’s disease (HD) family community plays in advancing scientific research. From their resilience in the face of this extremely challenging condition, to their extraordinary contributions to scientific studies, the HD family community continues to be an essential partner in driving progress toward treatments and, one day soon, a treatmentcure.
Participation in Research: A Commitment to Progress
One of the most inspiring aspects of the HD family community is their unwavering commitment to participating in research. Clinical trials, observational studies, and other data collection efforts all rely heavily on the active involvement of individuals with HD, their families, and at-risk individuals. These studies demand time, effort, and emotional resilience, yet the HD community consistently rises to the occasion, understanding that their participation is key to scientific progress.
This has been a long-standing theme in HD research. The Venezuela project began in 1979, a critical project in the history of HD research, enabled by the contributions of families from communities in this region. Their participation laid the groundwork for identifying the genetic mutation that causes HD, revolutionising the field and enabling genetic testing and targeted research.
Selfless contributions
Programs like Enroll-HD, a global observational study, owe much of their success to the dedication of HD families. More than 350 projects interrogating Enroll-HD data have been conducted, and more than 150 peer-reviewed manuscripts detailing their finds have been published – a huge achievement!
Additionally, genome-wide association studies (also called GWAS) have been made possible through the donation of DNA samples from HD families to a variety of different registries. GWAS look to try and find genetic modifiers of disease; other genetic factors which track with symptoms starting earlier or later in life than might be predicted based on CAG number alone. Many of these newly identified modifiers from GWAS of people with HD are now top priority targets for drug discovery by academic and industry researchers alike.
Biomarker identification and tracking studies, such as HD-Clarity and iNFLuence-HD, rely on the collection of cerebrospinal fluid (CSF) samples and blood samples to identify measurable indicators of disease progression. Similarly, many stem cell and brain cell studies, which transform donated skin cell samples into disease models, have been a direct result of the generosity of HD families.
More and more HD research studies are using donated human brain samples. This tracks with the advent of many fancy new techniques that allow researchers to examine post-mortem brains at a cell-by-cell level, increasing the amount of information gathered from these precious samples by orders of magnitude. The findings that come from those studies get us closer to understanding HD in people, and closer to a treatment.
Sharing Personal Stories: Humanising the Science
The HD family community has been instrumental in humanising the science behind the disease. By sharing personal stories through blogs, conference presentations, and other outlets, they provide researchers, policymakers, and the general public with a window into the real-life impact of HD. These narratives inspire scientists to work harder, inform the design of patient-centred clinical trials, and help ensure that new therapies address the true needs of those affected.
Projects like My HD Story and POWER-HD further enrich the research landscape by collecting HD family member-reported outcomes, details of peoples real-world lived experiences, and other vital data. Moreover, these stories foster empathy and understanding, breaking down barriers of stigma and isolation. They serve as a reminder that behind every dataset and lab experiment are real people – families who are fighting to ensure a better future for their loved ones and others.
The Power of Advocacy and Awareness
The HD community has been a relentless force in advocating for increased funding, research opportunities, more social supports, and greater public awareness. Many of the local HD organisations around the world are driven by families and caregivers, and have worked tirelessly to bring HD into the spotlight. These efforts have not only fostered public understanding but have also paved the way for governments and private entities to prioritise HD research initiatives. Without their advocacy, many breakthroughs in funding and resources would not have been possible.
Notably, HD advocates have brought their voices to regulatory bodies like the FDA, ensuring that the patient perspective is central in evaluating new therapies. This advocacy has been crucial in shaping guidelines for drug development and approval processes, making them more responsive to the needs of the HD community.
A Call for Continued Partnership
As we give thanks for the HD family community, it is also a call to action for researchers, policymakers, and society at large to continue prioritising their voices and contributions. Building on this partnership means:
Designing studies that respect and prioritise the needs of participants.
Ensuring that research findings are communicated back to the community in a timely and accessible manner.
Advocating for continued investment in HD research and community resources.
Looking Ahead with Gratitude
Progress in HD research would not be possible without the courage, generosity, and perseverance of the HD family community. They are the heart and soul of the fight against Huntington’s disease, and their contributions illuminate the path toward a future free of this devastating condition.
As we reflect on all that has been achieved, let us reaffirm our gratitude to this remarkable community and recommit to working together toward our shared vision of hope, healing, and discovery. So this Giving Tuesday, we encourage everyone from the HD community, particularly those from HD families, to look inward, acknowledge your contributions, and give yourself some gratitude.
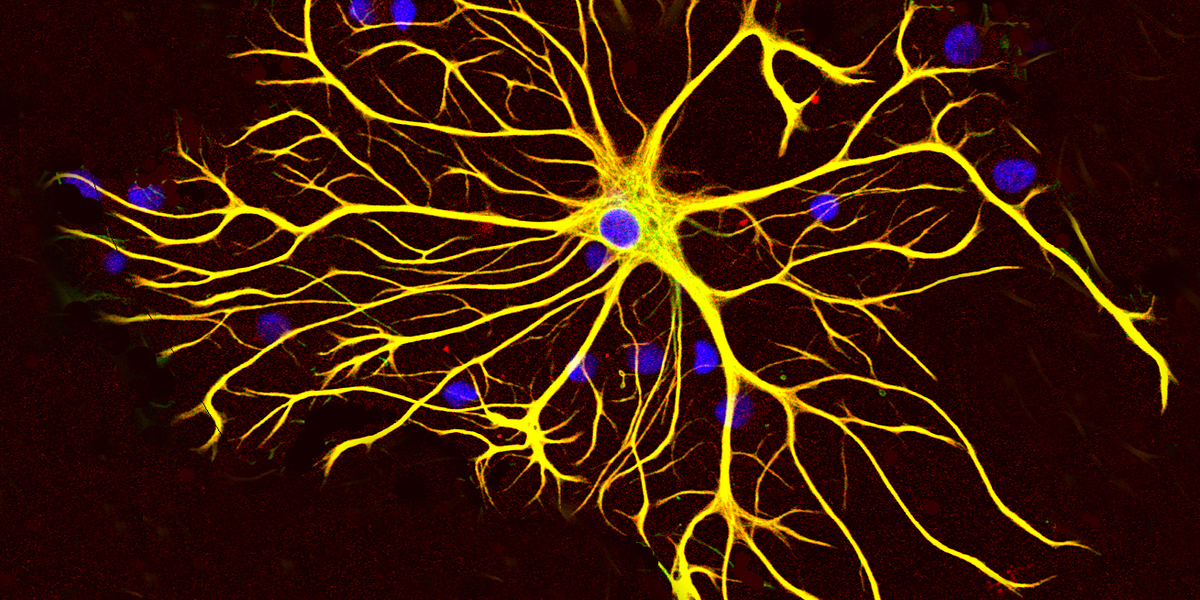
A new study led by researchers from Columbia University used postmortem brain samples to show that a special type of brain cell, called astrocytes, may play a role in how certain nerve cells are lost in Huntington’s disease (HD). This could have important implications for how we understand disease progression as well as for the development of new therapeutics that could target these cells specifically.
Star-shaped astrocytes are important for brain health
Nerve cells are responsible for sending signals across the brain that control our behaviour, mood, and help with communication between brain and body. These cells are the most affected in HD and are slowly lost as the disease progresses.
Besides nerve cells, our brain is made up of several other types of cells, including astrocytes. Astrocytes play an important role supporting nerve cell health and help with information processing.
Like nerve cells, and actually most cell types in our bodies, astrocytes also have the huntingtin gene switched on. In people with HD, this means they make the toxic huntingtin protein.
Several studies have indicated that this might cause astrocytes to not work as well in HD brains, including changes in how they interact with nerve cells. These changes are thought to contribute to HD progression. This new study sought to figure out exactly what changes are happening in astrocytes in HD.
Astrocytes: one size does not fit all
Researchers have long known that in people with HD, nerve cells in specific areas of the brain are more vulnerable to dying, a process called neurodegeneration. In this study, the authors looked at how genes are switched on or off in different regions of the brain using postmortem tissue. They found that differences in gene levels from healthy and HD donors were linked to specific brain regions and certain nerve cell types.
Across different brain regions, astrocytes are also not one uniform cell type. They are named for their star-like shape, and like the stars in the sky, there are many distinct types of astrocytes. These differ in shape, structure, and the roles they play in the brain, including how they interact with other cells. The researchers in this study identified several astrocyte subgroups in brain regions that are either vulnerable or resilient to neurodegeneration in HD.
Interestingly, a subgroup of astrocytes were present in HD brains, but not unaffected control brains in a region called the caudate nucleus, an area where nerve cell death is especially high in HD. This highlights that not only nerve cells are affected in vulnerable brain regions, but also that astrocyte subgroups are changed.
The presence of this subgroup of astrocytes meant that astrocytes that have specific functions were replaced, which could change how they interact with nerve cells in this region. Looking at different stages of disease progression, certain clusters of astrocytes were either increased or reduced. This could affect how these astrocytes might support nerve cells or maybe even contribute to their loss.
How do changes in astrocyte functions affect nerve cells?
Astrocytes have many different roles, but one of the most important ones is in brain metabolism. These cells help manage and process nutrients that nerve cells need to stay healthy and to function properly. These nutrients include different types of sugars, cholesterols, and fats.
There are many different kinds of fats and fat-like substances, also called lipids, in the brain. Previous research studies have identified changes in the amount of specific types of fats in HD brains compared to controls. The researchers in this study found that the amount of several types of fats tracked with disease severity in HD brain tissue.
But how do different lipids affect nerve cells? To test this, healthy nerve cells were grown in a dish and exposed to a stressor alongside the specific lipids identified in the study. In the context of cell stress, these appeared to be toxic and caused nerve cells to die.
An outstanding question is whether and how astrocytes contribute to the changes in lipid expression. It is not yet known whether astrocytes play a role in regulating those specific lipids. However, as astrocytes are the cell type that takes up and secretes the majority of lipids, it is important that future research establishes whether astrocytes contribute to nerve cell loss through changes in lipid metabolism.
Astrocytes: good guy or bad guy?
This study found a specific type of astrocyte that there are lots of in brain regions less affected by HD but very little of in the most affected areas of the brain. They found that this type of astrocyte has a particular group of genes switched on more than normal. These genes encode a type of protein called metallotheioneins.
The metallotheionein proteins help to protect cells from a damaging type of stress, called oxidative stress. This type of stress is caused by an imbalance of ‘bad’ reactive molecules and ‘good’ antioxidants. Increased levels of oxidative stress have been reported in HD and are known to damage cells.
Astrocytes are thought to play a key role in protecting nerve cells from damage caused by oxidative stress. The researchers in this study identified a specific gene, metallotheionein-3, which was associated with a neuroprotective astrocyte subgroup. When nerve cells were exposed to toxins in the laboratory, astrocytes that expressed higher levels of this gene could protect these nerve cells from cell death.
A new modifier of HD?
In HD, the age of disease onset generally tracks with the number of CAG repeats in the huntingtin gene, where more CAG repeats lead to an earlier age of onset. However, folks with the same CAG number can get symptoms earlier or late in life. This is partly due to genetic modifiers; small changes to the DNA letter code in other genes that can also affect the age of onset of HD symptoms.
In this study, researchers looked across 390 people with HD to see whether they could find genetic signatures in the metallotheionein-3 gene that tracked with the age when symptoms first appear.
Three small genetic signatures appeared to be linked to a later onset of symptoms in people with HD, whilst two other genetic changes appeared to increase how much this gene is switched on in a specific brain region, the prefrontal cortex. This highlights the potential clinical relevance of this gene and may represent a new way to target astrocytes therapeutically by designing drugs to change the expression of the metallotheionein-3 protein.
Astrocytes – a potential therapeutic target in HD?
The majority of cells lost in HD are nerve cells, but from studies like this, we are learning more about how other cell types and their functions are affected in the HD brain. The brain is incredibly complex, and by understanding more about other cells like astrocytes, we also learn more about how changes in cell-cell interactions may lead to neurodegeneration.
Unravelling the intricate relationships between nerve cells and astrocytes could be essential for developing effective therapies for HD. I like to think of the brain as an orchestra, where all instruments need to play well together. As such, it’s not sufficient to just target one part such as nerve cells and therapeutics need to target all cells affected by HD.
A lot of the findings from this study rely on human postmortem brain tissue and would not have been possible without organ donation. It’s the generous and selfless contribution that individuals make, that allows research like this to be possible.
TRIGGER WARNING:This article contains a frank discussion around the challenges and realities of living with Huntington’s disease, as well as caring for those affected by it. Topics include suicidal ideation, threats to family members, financial distress, paranoia, severe anxiety, feelings of hopelessness, and loss of identity. We understand that this may be a difficult article for some to read and caution people about reading this article who may not be in an appropriate headspace to think about such topics. While these types of conversations are difficult, they are necessary to inform those who are unfamiliar with the disease, to help them try to understand the stark realities of living with Huntington’s, and the devastation that it causes for those from affected families.
The regulatory agency in the US that approves all medications, the Food and Drug Administration (FDA), considers disease severity and availability of other treatments throughout the approval process. FDA representatives participate in meetings with those living with the diseases to hear their lived experiences. This can be critical for advancing therapeutics in a timely manner that meet the needs of a patient population. On November 13, 2024, such a meeting took place in College Park, Maryland, bringing people living with Huntington’s disease (HD) and their caregivers face-to-face with the FDA.
The opportunity
The Huntington’s Disease Society of America (HDSA) coordinated the Externally-led Patient Focused Drug Development (EL-PFDD) Meeting between HD families and the FDA. HDSA organized a similar meeting in 2015, but a lot has changed in the past 9 years – research has taken great strides, we’ve gone from having one disease-targeting clinical trial to having many, and there are dozens of pharmaceutical and biotechnology companies interested in advancing drugs for HD.
The 2024 meeting was described as “a one-time opportunity to provide the FDA and other key stakeholders, including medical product developers, health care providers, and federal partners, your perspectives on the symptoms that matter most to you, impact the disease has on your daily life, and your experiences with currently available treatments”. This was a once-in-a-lifetime chance for many to be heard by the agency that has the chance to approve medicines for people living with HD.
The goals
The goals for the meeting were to educate, inform, and advise the FDA and medical product developers on the challenges of living with HD and advocating for disease modifying drugs. The impact of living with HD at pre-symptomatic, early, and mid stages was shared.
The agency was informed about the treatment outcomes that the community prefers and the risks that they’re willing to take to get treatments for HD. Advice was given so that the FDA could understand the challenges people with HD face with current clinical trial participation and how it could be improved.
The structure
Following opening remarks and a clinical overview, the day was divided into two panel discussions, one on health effects and daily impacts and another on current approaches to treatments.
People living with pre-symptomatic, early, and mid-stage HD shared their stories and experiences along with caregivers. Together, these perspectives and the information collected in this meeting will be used to “inform FDA’s decisions and oversight both during drug development and during the review of a marketing application to treat HD”. Put simply, teaching the FDA what it’s really like to live with HD could have a big impact on advancing treatments for HD.
After the personal statements, there was a large group facilitated discussion using specific survey questions that were distributed to the HD community by the HDSA ahead of this meeting. This survey, on HD Symptom and Treatment Impact, will be open until December 31, 2024 and the HDSA expects to have results from that survey available in February of 2025. At the event, various HD family members were asked to share their lived experience with the FDA as it related to the question in the survey.
Opening remarks:
Dr. Arik Johnson, Interim CEO and Chief Mission Officer, HDSA
The morning opened with a welcome message from Dr. Arik Johnson, thanking everyone for participating in this important meeting – both those from HD families for sharing their stories and the FDA for listening. There were over 60 people living with HD in the room and over 140 people who registered to attend virtually from across the globe, from 43 US states and 8 countries. The HD community was ready to be heard!
Arik said, “The time is now, for this meeting and for this opportunity”. There is more research happening now at earlier disease stages. As trials shift to testing drugs at earlier disease stages, there are different outcomes and risks, of which regulatory agencies and product developers need to be aware.
Dr. Teresa Buracchio, Director, Office of Neuroscience, Center for Drug Evaluation and Research, FDA
Dr. Teresa Buracchio’s job at the FDA is to oversee drugs in development for a variety of neurological conditions, including HD. She assured the crowd that many people across the FDA are listening, and that the FDA takes these meetings very seriously. She noted that the people living with HD and their caregivers are the experts, and the FDA is eager to listen to their stories. Teresa said, “the voice of the patient is very important to us”, and the FDA references these reports when working to advance drugs for diseases.
It is the job of the FDA to ensure that the benefits of therapeutics outweigh the risks. So understanding how patients view the risks and benefits of treatments will help them advance treatments that will have a positive impact on people with HD. Teresa noted the exciting advancements that have been made for Alzheimer’s disease and genetic forms of ALS and she said they’re excited to incorporate similar advances to the field of HD.
Dr. Victor Sung, Professor of Neurology, Division of Movement Disorders and Director of the HDSA HD Center of Excellence, University of Alabama
Dr. Victor Sung gave a clinical overview of HD, sharing details about the natural history and progression of HD. He noted that HD is considered a rare disease and is less well known than other diseases. Even still, the prevalence of HD is actually similar to ALS, which gets a lot more “press time” and is more recognizable by name.
Victor detailed the genetics of the disease; that it is caused by the repetition of a C-A-G letter code in the huntingtin gene that’s on the 4th chromosome. But he also highlighted the socioeconomic burdens of HD and how it differs from other diseases. HD impacts wage earnings from people affected by the diseases as well as caregivers, generation after generation. It also differs from other brain diseases that are sporadic, like ALS, in that it can impact many people within a family, sometimes entire generations. The details that Victor shared demonstrated to the FDA that the tragedy of HD is truly unparalleled.
He then showed data that suggests there are changes occurring before people start to show symptoms of HD, like behavioral and cognitive changes associated with thinking, learning, and memory. This suggests developing early treatments for HD is potentially feasible.
He ended by saying that we have no disease modifying treatments for HD right now, but we are pushing into that realm. “The future is bright. There are lots of things happening in the disease-modifying space for HD. But we can only do this together, and we will do this together.”
Panel 1: Health effects and daily impacts
The first panel began with representatives at the pre-symptomatic, early, and mid stages, as well as a family member. They shared their stories about living with HD at each of these stages, highlighting how the disease, or knowledge that they’ll develop the disease without some sort of intervention, has affected their lives.
Speakers underscored the impact that HD has had, influencing the decision to have biological children, creating early conversations around life insurance and retirement planning, and becoming a caregiver to parents who represent a constant reminder of what the future holds unless we figure out a way to slow or stop HD.
Speakers detailed the changes they’ve experienced because of HD, such as a reduced ability to think through problems, multitask, and organize their lives, which has led to a loss of jobs, the ability to drive, and independence, causing an overall feeling of a loss of identity. Physical exercise and outdoor walks that used to be the highlight of a day now brought feelings of dread because of balance issues that have caused falls, leading to cuts so severe they require stitches. The emotional changes that HD brings were also discussed, with the onset of depression, anxiety, and panic disorder.
Panel 1: Group discussion around health effects and daily impacts
Questions that were discussed with the group from the HD Symptom and Treatment Impact survey around the health effects and daily impacts of living with HD were:
Of all the symptoms that you have experienced because of your condition, which 1-3 have had the most significant impact on your life?
Are there specific activities that are important to you, but that you cannot do at all or as fully as you would like because of your condition?
As it relates to your condition, what does a good day / bad day look like?
How has your condition changed over time?
What worries you most about your condition?
Pre-symptomatic
People living with pre-symptomatic HD said that cognitive and psychiatric symptoms are the primary issues, citing anxiety, depression, emotional outbursts, mind fog, and difficulty concentrating.
People shared that even at this stage they began to feel a loss of identity. Some people with high-level jobs, such as those working in finance on Wall Street, were fired because of changes experienced due to HD even 5 to 10 years before anyone on the outside would say they had symptoms.
The group noted that even though they are typically considered pre-symptomatic until HD-associated movement symptoms begin, there are real problems that begin to present in this stage. People overall felt like they were being forced to live their lives in a holding pattern, unable to receive treatment because they didn’t yet have symptoms, but also unable to receive treatment for the symptoms they were experiencing because they were being told they aren’t severe enough.
Early-stage
People living with early-stage HD cited some of the same issues as the pre-symptomatic group, highlighting problems with anxiety, depression, difficulty concentrating, and memory lapses. While the overarching issues were the same, the tone captured the more advanced state of these issues.
Overall, people cited that at the early-stage HD has caused them significant worry about the future. They fear becoming a burden to their loved ones and the impact that HD will have on their children. The thinking problems that increase during this stage cause people to make poor financial decisions and prevent them from being able to pay their own bills.
Participants stated that the early-stage brings changes related to independence – those who were once fully independent transition to being fully reliant on others for many basic functions, which is devastating, particularly for those who don’t have a support system. And for those seeking intimate relationships, HD has made dating “impossible”. An audience member with early-stage HD shared that they’re afraid to hold their grandchildren because of balance issues. Others shared that they avoid family functions because they don’t want to go in public or be seen for fear that people will think they’re drunk or on drugs.
This group also said emotional outbursts were a serious concern, becoming dangerous at times with the fear of law enforcement involvement. Others detailed the increase in paranoia at this stage, causing one person to hold a shotgun up against their own child because they didn’t know who they were or why they were there. Situations with law enforcement and nursing staff can easily become reactive because of such paranoia.
Mid-stage
People living with mid-stage HD shared that they’re unable to do work that they’ve done in the past or have been trained for, that they’ve lost independence, that they’re no longer able to drive, and that they’re financially unstable. The massive financial burden that HD causes was noted, both from the loss of income for the person living with HD as well as the loss of income from the caregiver rerouting their time to take care of the person with HD.
At the mid-stage, components of a good day related to things many of us often take for granted – a good night’s sleep, forgiving yourself for missteps and realizing it’s okay to not be perfect, and not having urinary incontinence. Those in this group cited bad days as those fraught with obsession over death and suicidal ideations.
For people in the mid-stage of HD, participants cited that symptoms around clumsiness, movements, anxiety, and difficulty concentrating have had the biggest impact on their lives. They shared that their ability to walk and speak has degraded to the point of constantly stumbling and having slurred speech, which causes them to feel uncomfortable in social situations and lose social connections and peer support. Gastrointestinal issues, which are a known problem for many people living with HD, were brought up for the first time in this group.
For people in mid-stage HD, many were most worried about the capacities they’re losing, or will lose next. They shared grief over loved ones constantly losing pieces of themselves. Caregivers expressed worry over what will happen when loved ones need more consistent full-time care and the financial impact that will have on their families.
Panel 2: Current approaches to treatment
The second panel centered around current approaches to treatment, bringing in a different group of panelists, one each with pre-symptomatic, early, and mid-stage HD, along with a family member of a person living with pre-symptomatic HD.
Panelists shared that the psychological and physical toll of HD is grueling. People with pre-symptomatic HD want to participate in trials to prevent the onset of the most severe symptoms but are told their disease hasn’t progressed enough to be considered for trials. The frustration around wanting to try but not being able to is defeating. One panelist stated, “We know what happens when we do nothing. We just want a chance to fight.”
Those with multi-generational experience with HD shared stories of watching their parents use treatments for their movements, only to have their engagement with the world slowed. Panelists shared their experiences participating in clinical trials that were ultimately halted, saying that even though they felt they were benefiting they no longer had access to that medication. They also detailed the heart wrenching conversation with their children, having to explain the lack of access to a drug they thought helped.
A heartfelt plea was made to the FDA for a special designation for unproven and unapproved treatments that make people feel better regardless of trial outcome, to help develop better determinants for trial endpoints, to move trials into pre-symptomatic groups, to focus on cognitive, psychiatric, and behavioral symptoms, and to help increase funding for HD research.
Panel 2: Group discussion around current approaches to treatment
Questions that were discussed with the group from the HD Symptom and Treatment Impact survey around the current approaches to treatment for HD were:
What are you currently doing to help treat your HD symptoms?
How has your treatment regimen changed over time and why?
How well does your current treatment regimen treat the most significant symptoms of HD that you experience? How well do your treatments improve your ability to do specific activities?
How well have these treatments worked for you as your condition has changed over time?
What are the most significant downsides to your current treatments and how do they affect your daily life?
Short of a complete cure, what specific things are most important to you around delaying the progression of HD?
If you could have a reduction in symptoms, what would make the most positive change in your life?
Do you have any concerns around participating in a clinical trial?
Pre-symptomatic
At the pre-symptomatic stage, many people indicated that they’re taking medication for anxiety and depression. Tellingly, caregivers also stated they’re on similar medications along with blood pressure medication, suggestive of the ripple effect that HD has through families. Participants also cited non-medical treatments that have been suggested to slow disease onset, such as exercise, good sleep, a healthy diet, and a positive mindset.
By and large the most significant downside noted from the pre-symptomatic group was their inability to qualify for clinical trials. While they see massive changes in themselves that they believe are from HD, they’re constantly told they don’t have symptoms and thus don’t qualify for trials.
For people with pre-symptomatic HD, they expressed a strong desire to try anything in trials that could slow disease progression or delay onset. They noted that a treatment wouldn’t have to even hold the promise of a cure, and that delaying onset would be enough. People were most interested in medications that could potentially treat thinking changes brought about by HD, which could help them keep their jobs for longer to offset the financial burden associated with HD.
Early-stage
Along with anxiety and depression medication mentioned by the previous group, people living with early-stage HD stated that they’re prioritizing exercise, getting outside more, and participating in music therapy. People also said that they’re challenging themselves intellectually, by going back to school and playing brain games.
In the early-stage HD group, some felt that medications used to control movement symptoms have been critical. However, some of these medications don’t work for everyone, so trying several regulatory approved medications allowed them to find one that helped control their movements while also decreasing negative mental side effects that caused suicidal ideations. Others found that ADHD medication has helped with thinking and memory problems. One participant stated that medication that she takes for anger outbursts has allowed her to keep her job and has helped keep her family together. Several in this group cited medical marijuana as being a “game changer”, although laws vary state-by-state in the US, which some noted as being an issue for access and regulation.
Many people in the early-stage group noted challenges around trial participation related to traveling far distances and logistical difficulties for caregivers who often coordinate care and trial participation. Others detailed challenges with navigating the US healthcare system. Some HDSA Centers of Excellence don’t accept some insurance plans and some medications prescribed by physicians for HD symptoms aren’t covered by insurance.
As in the previous group, people living with early-stage HD stated that they want medications that could help with cognitive effects and thinking problems. This could help with time management issues, help people keep their jobs for longer, and defray financial concerns by extended wage-earning years. Improving cognition could also help people communicate better and maintain independence for as long as possible.
One person from the early-stage HD group cited that they just wanted to retain their dignity. Being able to go to the bathroom and take a shower on their own would have the most positive change in their life.
Mid-stage
From the mid-stage group, some cited that they’re taking sleep medication. One said that sleep apnea machines have helped with getting better rest. Others said they’re working to improve the small things that affect health, like dental, vision, and hearing problems, that can add up over time.
Others mentioned small lifestyle changes that have made a difference, such as always using a straw and moving to a single level house so that falls down stairs are less likely. Those in the mid-stage group stated they’re working to make accommodations for what they or their loved ones are experiencing, which can be as simple as putting a tablecloth down instead of trying to get someone to eat more neatly. Astutely, one participant noted that you have to think outside the box for HD to find things that will work at each stage.
One participant said that her life changed when her husband went on antipsychotics, which helped control abusive and paranoid behavior.
As for the previous groups, trial location was a concern. One participant from the mid-stage group said, “I will do anything if it means helping my husband and helping my kids”. HD is a family disease, evidenced by the caregivers that stood up, literally and figuratively, at the recent FDA meeting for their loved ones with HD. Another woman said she would give her life in a trial if she thought that meant it would secure the future for her daughter and granddaughter. A sentiment she said that she hopes doesn’t fall on deaf ears.
The conversation will continue
A representative from the FDA shared that they’re having a virtual patient listening event on December 4th to hear from the community on what they think about enrolling in a trial at pre and early symptomatic stages. She encouraged everyone to register, listen, then add their thoughts into the meeting notes. After the meeting they’re going to develop a summary to help inform the FDA and stakeholders in drug development. Registration for this event will close December 3, 2024.
Arik delivered closing remarks, thanking everyone who participated, both in person and online. He noted that we have a lot of work left to do and this is just the first step in the process. There is still a lot that wasn’t said, but the work will continue to ensure that everyone is heard. Every lived experience with HD, in every stage of HD, is impactful and matters, and will be used in future decision making.
Arik noted that there are things that can be done right now, such as participating in observational studies like ENROLL-HD, POWER-HD, and MyHDStory. He noted that it’s important for everyone to take care of themselves and reach out to people if you need help.
The takeaways
The discussions with the pre-symptomatic group highlights that the term “symptomatic” needs to be reconsidered, as the time when many are considered to be without symptoms is filled with behavioral and psychological changes. For them, the pre-symptomatic stage doesn’t mean they don’t have symptoms. People reported feeling frustrated that they see that they’re changing, with anxiety, depression, or executive functioning, but people don’t see that on the outside and doctors will tell them to come back in 5 or 10 years when they start to show symptoms.
At points, an FDA representative took the microphone to directly ask the audience what types of medications would be most useful for them. Almost everyone agreed that having something that works to improve cognition would be their number one choice. This could improve their thinking so that they could keep their jobs longer, defray the financial burden of HD, and help them communicate better with loved ones.
The direct exchange between the FDA and HD families underscored what this meeting was all about – a two-way conversation to help the FDA understand the needs of HD families to get this community treatment options as soon as possible to improve their lives. The HD families were heard.
Thank you!
Any meeting to educate the uninformed about HD will be emotional. There’s no way to describe HD without stirring feelings of loss, grief, and despair. Even through that though, a strong, bright thread remained – that of resilience, that of hope, that of determination.
Many experiences were punctuated with statements around wanting to not just survive but to live, wanting to participate in trials, wanting to be a part of the science that will get us a treatment for HD. Along with the heartache that HD brings, there’s no doubt that the FDA also heard the underlying message of strength within this community.
To everyone from the community who participated, attended, and shared your stories – thank you. Thank you for your willingness to be vulnerable. Thank you for your honesty. Thank you for standing up to change how the FDA views HD. You represented every HD family member who couldn’t be in that room, and you did it gracefully. Because of you, the needle was moved today. Because of you, we’re one step closer. Because of you, the HD community stood face-to-face with the change makers and was heard. Thank you.